审核作业:
4.5.1 审核小组由管理者代表指派合格的审核员担任。
4.5.2 管理者代表依据年度审核计划于三天前以《QMS及ISO22000体系审核通知单》通知被审核单位审核时间及项目,受审单位对审核时间若有异议时由管理者代表更改。审核的范围应该包括: 品质及ISO22000体系及其记录、偏差和产品处置及确认CCP受到控制等的评审。
4.5.3审核会议:本会议由审核小组视实际情况可召开或不召开,如需召开则由管理者代表于会议时介绍双方成员、审核项目及审核行程等事项。
4.5.4现场审核:由审核小组依据《内部审核通知单》或审核前会议的工作分配执行审核,并将审核结果记录于《内部审核查检表》,审核的缺点记录于《内部审核不符合报告》,现场审核时被审核单位可派员随同解说,若有缺失则每一缺失由审核人员开一张《内部审核不符合报告》。
4.5.5 审核末次会议:本会议得由审核小组视实际情况召开或不召开,如需召开则由管理者代表负责召集双方人员,由审核小组提报审核的缺失,被审核单位如对审核结果有异议时,可当场提出说明或证据资料,经由管理者代表审核判定,如未召开本会议,则被审核单位于收到《内部审核不符合报告》三天内,需提出对有异议内容的书面说明资料送管理者代表审核判定,如未提出则视为完全承认。
4.5.6 审核记录撰写、汇整及审核:审核小组于审核完毕后应于当天填妥《内部审核不符合报告》交管理者代表汇整审核,然后由管理者代表汇总为《内部审核总报告》提交总裁审核并在管理审核会议上报告,以达到高层管理人员对公司QMS及ISO22000体系运作状况的了解。
4.5.7 缺点的判定:
4.5.7.1主要缺点:
A.导致体系功能性及系统性失败的缺点;
B.相同的轻微不符合事项,在多个部门重复出现而导致QMS及ISO22000体系失效。
C.违反国家强制性法规的.
4.5.7.2次要缺点:除上述主要缺点的事项外,其余因执行不彻底或偶发的疏忽所造成的缺失则判为次要缺点。
4.5.7.3观察事项:纠正措施实施中或所依据的公司QMS及ISO22000体系未能符合相关质量的标准,需长时间改进者(改进时间**过二个月)由审核员判定为观察事项,但仍需开立《内部审核不符合报告》要求改进并列入跟催项目进行管制。
4.5.7.4建议事项:公司QMS及ISO22000体系规定符合相关质量标准但仍可透过建议须达到改进目的。
4.5.8由审核小组发出的《内部审核不符合报告》,由管理者代表予以统一编号跟催,审核执行完毕后由管理者代表在《年度审核计划》中直接划记结案。4.5.9缺失改进及审核:被审核单位依《内部审核不符合报告》所规定期限及其内容提出改进计划,缺失改进后由管理者代表指派审核小组人员作效果的评价,如改进后仍有缺失,则应持续改进至有效为止。

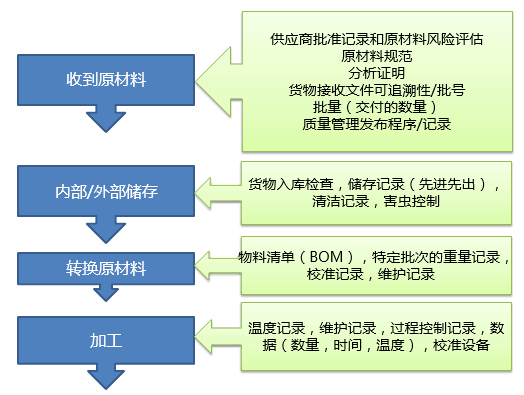
产品的测量和监控
4.2.1质量部负责编制各类检测规范、《HACCP计划》,明确检测点,检测频率,抽样方案,检测项目,检测方法,判别依据,使用的检测设备等。
4.2.2进货检验:
4.2.2.1对生产购进物资,仓库保管员核对送货单,确认物料品名、规格、数量等无误,包装无损后,置于待检区,填写《收货单》,交QC检验员。
4.2.2.2QC检验员根据《检验作业规范》进行对来料检验记录,并填写《检验报告》。
a)仓库根据合格记录或标识入库手续;
b)验证不合格时,按《不合格品控制程序》进行处理;
4.2.2.3紧急放行:
原料、辅料、与食品接触材料不得紧急放行和让步放行。其他采购品紧急放行和让步放行时,应:
a) 由使用部门填写《紧急放行申请单》,经仓库及品检负责人确认,总经理批准后方可执行。但必须抽取样品封存或送检。
b) 任何紧急放行的产品必须具备可靠的可追溯性,以便发现问题后能有效地追回。应在放行的产品上或后续的记录上加上“紧急放行”标识。
c) 在紧急放行的同时,检验员应继续完成该批产品的检验;不合格时应负责对该批紧急放行产品进行追踪处理。
4.2.2.4采购产品的验证方式
验证方式可包括检验、测量、观察、工艺、验证、提供合格证明文件等方式。经质量部主管审核批准的,可根据品质情况确定,**。对于质量部发出的**的物料,可不做标识。
4.2.3半成品的测量和监控:
4.2.3.1过程检验
对设置检验点的工序,检验员依据检验规程进行检验,合格交下一工序,并填写记录表,对于不合格品执行《不合格品控制程序》。
4.2.3.2巡回监控
生产过程中,生产班长(检验员)应对操作工人的自检进行监督,认真检查操作者的作业方法;使用设备等是否正确,根据需要进行抽检,并将结果反馈给操作者,发现不合格品应执行《不合格品控制程序》。
4.2.3.3半成品检验中,发现不合格品率接近组织规定值时,检验员应根据情况及时通知操作者注意加强控制;当不合格品率**过组织规定值时,应发出《纠正和预防措施处理报告》,执行《纠正与预防措施程序》。
4.2.3.4在所要求的检验和试验完成或必须的报告收到前不得将产品放行。如果因生产急需来不及检验而例外放行,应参照4.2.2.3的有关规定。

5.4 操作性前提方案
5.4.1根据前提方案的输入要求,应在如下方面分别建立操作性前提方案:
a) 与食品接触或与食品接触表面接触的水和冰的安全;
b) 防止交叉污染;
c) 有毒化学物质的标记、储存和使用;
d) 人员卫生;
e) 清洁和消毒:包括与食品接触的表面(包括设备、手套、工作服)的清洁度,手的清洗与消毒,厕所设施的维护与卫生的保持;
f) 虫害控制;
g) 交叉污染的预防措施;
h) 包装程序;
i) 对采购材料(如原料、辅料、化学品)、供给(水、空气、蒸汽、冰等)、清理(如废弃物和排水系统)和产品处理(如贮存和运输)的管理;
j) 生产加工和检验的作业过程。
5.4.2 每个操作性前提方案均应包括控制措施,明确如下内容:
a) 目的和范围:说明该操作性前提方案对哪些已确定的食品安全危害进行控制(包括适用的终产品、场所、工艺和危害类型)。
b) 职责和运行:说明该操作性前提方案及其控制措施的过程是如何进行的,应过程职责和权限的细节。
c) 监视:规定能够证实操作性前提方案(OPRP(s))有效的相关监视程序(参数、频率和记录要求)。
d) 纠正和纠正措施:当监视显示控制措施不适用时应采取的纠正和纠正措施。
5.4.3 操作性前提方案的验证:
a) 目的:验证操作性前提方案的有效性。
b) 方法:检查监视记录,验证方案是否得到实施;运行操作性前提方案,检查作业人员是否遵守规范,设备是否运转正常,仪器是否得到校准,以确定其控制的食品安全危害水平是否在预期范围内。
c) 频率:运行或变更后重新运行时和不**过六个月的时间间隔进行。进行。
d) 职责:由HACCP小组负责。
e) 记录:参加人员应填写验证签到表(会议签到表),编写操作性前提方案验证报告记录验证过程和结果。对验证不合格开立文件更改申请表或不合格处理单以对其进行纠正。
5.4.4 操作性PRP(s)应与组织的经营规模和类型,以及生产和(或)处理的产品性质相适应;无论是整体应用,还是用于特定产品或生产线,操作性PRP(s)应在整个生产体系中实施。

-/gbafcjj/-
http://iso9001fsc1.b2b168.com